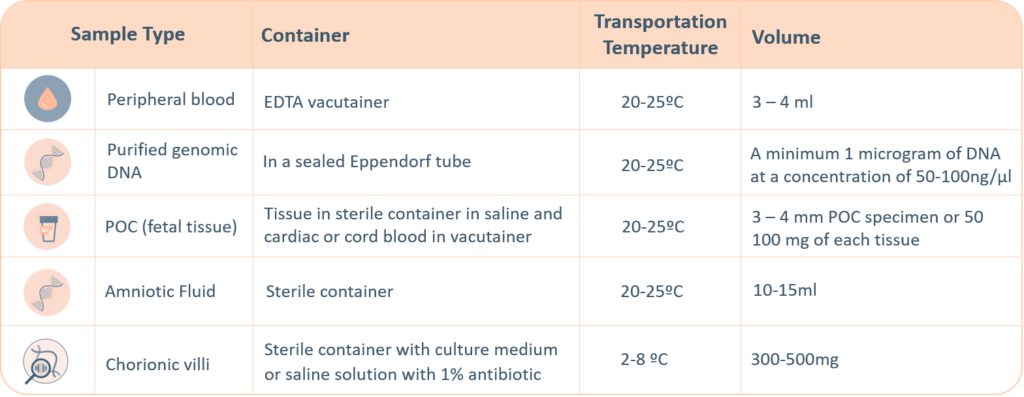
CMA Sample Requirements
The following sample types are accepted for Igenomix genetic tests. A thorough labelling of the tube with unique identifying information is suggested, incorrect labelling can lead to rejection of the sample. The minimum required information to identify and accept a sample is – Patient’s full name, Date of birth, Gender and Medical Record Number.
- Maternal blood sample must be sent with all products of conception, CVS and Amnio samples.
- Precedence will be given to all prenatal samples.
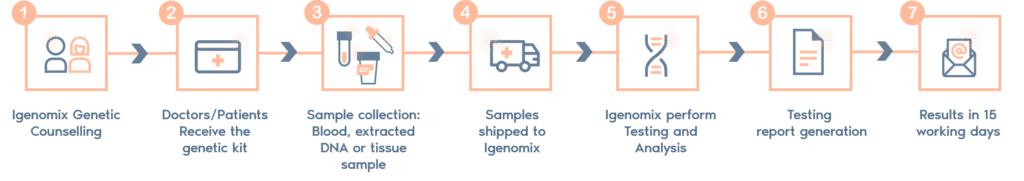
The ‘informed consent’ form and the ‘test requisition from’ (included within the provided kit) must be properly filled-in and signed by the patient and sent with the samples inside the shipping box or by e-mail to the laboratory. Igenomix will send you all the documents needed for the pick-up and transportation of the appropriate kit to our laboratory .